Xian Mallory
Department of Computer Science, Florida State University

I am a computer scientist who is interested in developing novel computational techniques for analyzing large data sets in order to understand biological phenomena and processes, with particular emphasis on cancer. My work draws upon methodologies from combinatorial optimization, statistical inference, and machine learning.
I graduated in May 2018 from the Computer Science Department at Rice University and am currently a postdoc at the same institute but with a slightly different research focus. My PhD and postdoctoral advisor is Dr. Luay Nakhleh. During my postdoc, I collaborate extensively with Dr. Nicholas Navin on developing machine learning tools to decipher cancer cell evolution. I also gained extensive experience in analyzing Next Generation Sequencing data through the collaboration with Dr. Ken Chen in MD Anderson during my PhD.
My research focuses on developing machine learning tools to detect structural variations in DNA using (the combination of) a variety of sequencing technologies, including Illumina, PacBio, Optical Maps and 10x etc, and to analyze single cell sequencing and deconvolve cancer cell heterogeneity. I developed several structural variation detection/characterization tools including HySA, OMIndel and BreakDown. I also participated in developing genomic tools such as BreakDancer, TIGRA-SV, CREST, novoBreak, BreakpointSurveyor and GMT Modeling. During 2015-2018, I was a member of Human Genome Structural Variation Consortium (HGSVC), where I took the leadership in developing and applying machine learning tools to newly sequenced data.
For my up-to-date CV, click here.
I develop tools to detect structural variations in human genomes via different sequencing technologies. Structural variations are variations in DNA occurring to more than 50 bases. It is called "structural" because variations occurring to a large number of bases may change the structure of protein. Detecting structural variations is an important bioinformatics topic because these variations may lead to genetic diseases such as cancer. I devoted near a decade of time to this area and here I list a few tools that I developed or participated in the development.
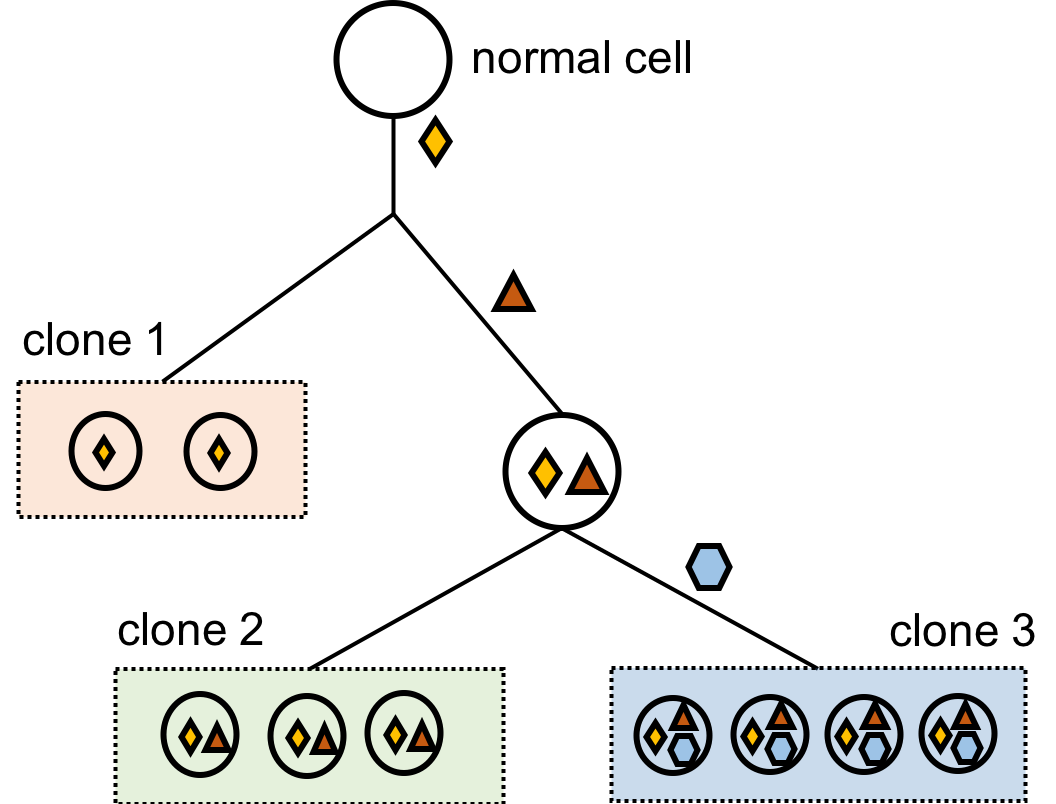
Cancer is by nature heterogeneous. After a normal cell gains a mutation (changes on DNA that may lead to cancer, denoted as the yellow diamond in this figure), some of its daughter cells may gain additional mutations (denoted as the red triangle) whereas some others don't. Such process continues until the doctors sequence the patients' genome, which can be understood as taking a screenshot of the patient's DNA. Since the evolutionary tree has different branches, each having their own signatures of mutations, the screenshot also contains multiple clones of cancerous cells. My research goal as a researcher in bioinformatics and computational biology is to characterize all subclones in the cancer and to recover the evolutionary history. There have been two ways to tackle this problem, and they both depend on the data given.
The first approach is by deconvolving the mixture of subclones from bulk sequencing data, where all subclones have been mixed together. BreakDown attempts to analyze such data by analyzing the variant allele fraction (VAF) of structural variations. Since mutations from the same clone have the same VAF, and the VAF of the mutations occuring to a parent clone is the sum of those at the daughter clones, it is possible to cluster all mutations by their VAFs and infer the number of clones, the evolutionary history and placing the mutations on the edges of the tree. Traditionally the VAF of single nucleotide variants is used for such clustering. However, in BreakDown, my colleagues and I found that strucutral variation is more suitable for such a task due to that it involves more genomic fragments and therefore is more sensitive to small clones. We published this paper on BMC Bioinformatics in 2014, and we noticed a recent citation by Nature Communications (11:730, 2020) that discussed BreakDown in detail.
The second approach is by inferring the mutations occurring on single cells and the corresponding evolutionary history from the cells. Single-cell sequencing is a relatively newer sequencing technology, which unlike bulk sequencing, can sequence one cell at a time. Such capability to separate the cells provides the chance to detect mutations on each cell. Suppose the cells represent the leaves on a tree, one can then infer the evolutionary tree based on the mutation profiles on the leaves. Quite a few studies have been done on using single nucleotide variants to recover the tree (see SCITE, OncoNEM, Sifit, SiCloneFit, SCG and BEAM). Their biggiest difference is the model the tool is under. The models include infinit-site model (assuming parsimonious), finite-site model (allowing back mutation, parallel mutation, multiple mutations on the same site) and in between these two are Dollo model. Few studies have been done on using copy number aberrations (CNA) to infer the evolutionary history of cancer. Since CNA played an important role in cancer progression, my work during postdoc focuses on CNAs by single-cell sequencing data.
The followings give more details on my work for each approach to decipher cancer heterogeneity.
600 W College Ave, Tallahassee, FL 32306
(email) xfan2@fsu.edu; xfan@cs.fsu.edu